
Molecular Static Calculations for Stacking Fault Energy in Alloys
Published:
Stacking fault energies (SFEs) are critical properties influencing the formation of growth twin boundaries in materials. Documenting SFEs is notoriously challenging due to their sensitivity to compositional changes and the intensive labor required for accurate measurement. While molecular statics (MS) and density functional theory (DFT) serve as computational alternatives for estimating SFE, DFT’s applicability is curtailed by its atom count limitations, introducing potential biases in multi-element alloys. Our approach leverages MS calculations with the LAMMPS software to determine SFE for a diverse range of alloy compositions.
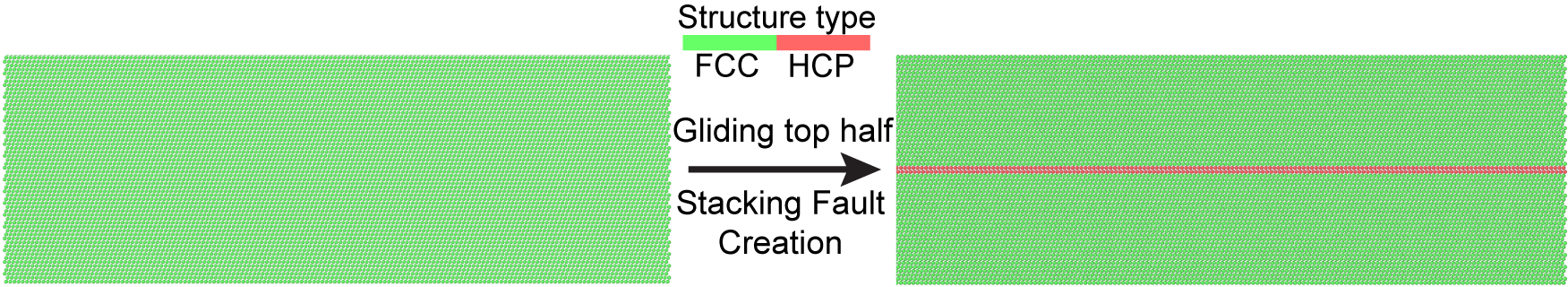
In these calculations, a perfect stacking fault is manually introduced by displacing the top half of an FCC structure along the <112> sliding direction, altering the stacking sequence from ABCABCABC to ABCBCABC. This fault introduces a local HCP structure, which is visualized using OVITO and Common Neighbor Analysis (CNA).
The formula for SFE calculation is presented as:
\[\begin{equation*} \frac{E_f - E_i}{A} \end{equation*}\]Here, \(E_i\) and \(E_f\) are the initial and final system energies, respectively, and \(A\) is the area of the gliding plane. The calculated SFE plays a pivotal role in advancing our knowledge of material behavior, aiding in the tailoring of material properties for specific applications. This is due to the direct impact of SFE on the formation of microstructures and the resulting mechanical properties, which highlights its fundamental importance in the field of materials science.